Evolution in the North American Basin In the 1990s, we began a NSF supported program of research to measure population genetic structure in deep-sea mollusks. This was the first concerted effort to study the genetic basis of population differentiation in the deep sea, apart from hydrothermal vents. A major breakthrough was our development of molecular genetic techniques to work with formalin-fixed small (<1mm) invertebrates. These new genetic methods made it possible to use extensive available collections of deep-sea species to explore the evolutionary-historical basis of deep-sea biodiversity on global scales, and added a new dimension to the use of museum collections in general for spatial and temporal analyses of population structure. Using these techniques, we quantified the population genetic structure of several molluscan species arrayed along a depth gradient in the western North Atlantic (see Fig.1). Genetic divergence among populations decreased with depth suggesting that the potential for population differentiation and speciation varied bathymetrically (Fig. 2a). The greater divergence for more shallow species was also apparent when we expanded the geographic scale to the entire Atlantic. Levels of genetic divergence were considerably lower for an abyssal pan-Atlantic protobranch bivalve compared to a bathyal species analyzed across similar geographic scales (Fig. 2b). For both species, bathymetric divergence occurred on much smaller scales than geographic. Genetic divergence among populations separated by 3 km in depth was similar to populations at the same depth but separated by 12000 km in distance! Isolation by depth appears to be considerably more important than isolation by distance in the deep sea. Patterns of genetic variation also indicated that deep-sea macrofauna could have strong population structure over small spatial scales (Fig. 3), despite the lack of obvious isolating mechanisms. Genetic divergence was sufficiently large for some species that they probably represent cryptic species complexes. There were no obvious oceanographic or topographic features that would impede gene flow at these depths. For example, simulated dispersal of neutrally buoyant passive particles released at depth in this region of the North Atlantic follow extremely complex trajectories (Fig. 4) suggesting larval exchange among depths should be possible. The small scale over which divergence emerges, and the lack of obvious oceanographic or topographic features that could impede gene flow, suggests divergence might reflect ecologically driven selection mediated by environmental correlates of the depth gradient. As inferred for numerous shallow-water species, environmental gradients that parallel changes in depth may play a key role in the genesis and adaptive radiation of the deep-water fauna.
We also quantified the genetic structure of several protobranch bivalves and gastropods at ocean-wide scales to test hypotheses about how major current and topographic features limit gene flow (Fig.s 5-7). Patterns of genetic variation differed for bathyal and abysssal species suggesting they are influenced differently by deep-water currents and the Mid-Atlantic Ridge. Our research is providing the genetic tools to explore population structure in the deep sea, and producing the first critical evidence of how and where evolutionary differentiation occurs in this vast and complex ecosystem. Below is a description of our ongoing research programs and questions. 1) Depth-Differentiation Hypothesis The deep sea is now known to be highly complex with strong geographic and bathymetric gradients that might influence the location, scales and dynamics of evolution. The rate of environmental change is a function of the rate of change in depth and proximity to coastal production. In the more steeply descending bathyal zone, depth parallels gradients of decreasing temperature, decreasing metabolic rates and increasing pressure. The bathyal zone is also a heterogeneous environment on large and small spatial scales. Not surprisingly, with strong vertical environmental gradients and a fragmented heterogeneous landscape, the bathyal zone supports high alpha and beta species diversity. The strong environmental gradients and greater biotic and abiotic heterogeneity at bathyal depths might impose different selective regimes that increase the probability of population differentiation and speciation. Because of its close proximity to land and coastal systems, the bathyal zone must have been strongly impacted by global climatic and oceanographic changes in the past. Variation in surface production caused by glacial cycles must have caused strong population fluctuations and bathymetric range displacement. The combination of paleoenvironmental change, fluctuations in populations size and the isolating effects of slope erosion during glaciation might have promoted population differentiation in the bathyal zone by both selective and non-selective mechanisms. The deeper, more remote, more uniform and topographically simpler environment of the abyssal plain is probably considerably less conducive to evolutionary divergence. In addition to the more pronounced spatial and temporal environmental heterogeneity at bathyal depths, several lines of evidence support the notion that this region promotes population differentiation and speciation. First, intraspecific population genetic divergence is greater at bathyal than abyssal depths for both mollusks and crustaceans. Second, phenotypic divergence as measured by multivariate morphological change in shell architecture in gastropods decreases exponentially with depth paralleling bathymetric patterns in genetic divergence. Third, recent evidence from paleontology, comparative phylogenetics, and molecular evolution suggest that geographic variation in evolutionary rates may play an important role in producing large-scale gradients in diversity. Similarly, depth-related variation in evolutionary rates might help explain why species diversity is greatest at bathyal depths. The intraspecific patterns of genetic variation within and among species, the morphological divergence and the correlation between evolutionary rates and species diversity all suggest the bathyal region may be an evolutionary hot spot for the genesis of the endemic deep-sea fauna. We are testing the hypothesis that population differentiation in deep-sea mollusks decrease with depth. This involves quantifying geographic and bathymetric patterns of variation in both nuclear and mitochondrial genes for a series of clams and snails along a depth gradient (500-5000m) in the western north Atlantic. 2) Forces Creating a Genetic Break at 3300m Our research has revealed a pronounced genetic separation between populations of the protobranch bivalves Deminucula atacellana and Neilonella salicensis above and below 3300 m. Populations on either side of 3300 m possess highly divergent haplotypes. The strong population divergence was unexpected and perplexing because there are no obvious oceanographic or topographic features that would limit gene flow between these regions (e.g. Fig 4). A similar break at 3300 m has been found for D. atacellana in the South Atlantic, although the data are more limited. Interestingly, these two protobranchs are not the only species to show a sharp break at 3300 m. In a global analysis of population structure in the cosmopolitan amphipod Eurythenes gryllus , France and Kocher (1996) found a similar pattern at virtually the same depth (Fig. 9). At abyssal depths, Atlantic and Pacific populations of E. gryllus were genetically homogeneous, but in both oceans, there was a pronounced divergence (16 S mtDNA) among populations above and below 3200m. The bathymetric divergence within ocean basins far exceeded that found between the Atlantic and Pacific populations at similar depths. These two species have extremely different life styles, natural histories, phylogenetic affinities and geographic distributions. Their congruent genetic divergence at 3300 m in vastly different regions of the world oceans, suggests that 3300m may represent a ubiquitous unrecognized phylogeographic barrier isolating organisms inhabiting different depth regimes. In our work we are exploring the forces that might produce such a sharp genetic discontinuity among multiple unrelated species.
General Sampling ProgramTo test the hypotheses outlined above, we took 28 epibenthic sled samples along a depth gradient from south of Cape Cod, Massachusetts to Bermuda (Fig. 1). The samples were taken evenly spaced from 1000-5000m depth along the transect shown in Fig. 1. We also sampled more intensively between 2500 and 3500m to test hypothesis 2 and at abyssal depths to test hypothesis 3. The transect parallels the Gay Head- Bermuda transect (GBT) of the Woods Hole Oceanographic sampling program of the 1960s to allow us to ultimately test temporal changes in haplotype frequencies of mtDNA.
|
Fig. 1. Numbers indicate location of the Woods Hole Oceanographic Institution's benthic samples (Sanders 1977) used in our previous genetic work. Solid line and abyssal circle indicate our recent sampling transect.
|
|||
Fig. 2a. Population divergence varies with depth. Relative genetic distances (UPGMA) among stations for each species. Station numbers are shown at branch tips and are depicted on the map in Fig. 1. The species from bathyal depths (yellow) exhibit much more population structure than do those from the lower bathyal and abyss (green ). From Etter et al. 2005. |
||||
Fig. 2b. Depth is more important than distance in fostering divergence. Genetic distance (pairwise FST) as a function of the geographical distance (km) and depth difference (m) separating all samples for a bathyal protobranch bivalve Deminucula atacellana and for the abyssal protobranch bivalve Ledella ultima. Both species have pan-Atlantic distributions and were sampled across similar scales. Distance-weighted least squares was used to smooth the surface. Only samples with greater than three individuals were included. From Etter et al. 2011. |
||||
Fig. 3a. Strong divergence occurs over small distances/depths. Multilocus (COI, 28S, CAL-intron) phylogenetic relationships among Neilonella salicensis at bathyal depths in the western North Atlantic. The 2 clades shown are likely cryptic species. From Glazier and Etter 2014. |
||||
Fig. 4. Simulated dispersal trajectories for neutrally buoyant particles released at 4 depths in the western North Atalantic. Trajectories are derived from a deep-ocean circulation model (FLAME) and are color coded based on time (red = 30 days, green = 180 days, blue = 360 days). From Etter and Bower 2015. |
||||
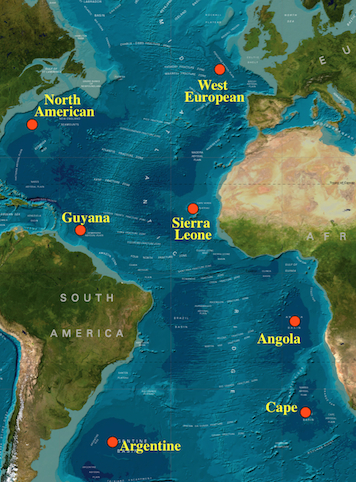
Fig. 5. Sampling locations to test ocean-wide patterns of population structure and the improtance of the mid-Atlantic ridge in isolating populations |
||||
Fig. 6. Neighbor-joining tree comparing genetic distance in the 16S rDNA gene among stations using Fst values. Although there was no difference between the eastern and western North Atlantic (across the mid-Atlanrtic ridge), populations at different depths did differ in both the North and South Atlantic. |
||||
Fig. 7. Neighbor-joining tree comparing genetic distance in the 16S rDNA gene among stations using Fst values. Station numbers are color coded based on whether they come from the eastern (green) or western (red) Atlantic. Populations were significantly different across the mid-Atlantic ridge. NAB = North American Basin, WEB = West European, Gy = Guyana, Ang = Angola, SL = Sierra Leone
|
||||
Fig. 8. Neighbor-joining tree comparing genetic distance in the CO1 gene among stations using Fst values. Station numbers are color coded based on whether they come from the eastern (green) or western (red) Atlantic. Populations were significantly different across the mid-Atlantic ridge. NAB = North American Basin, WEB = West European, MED = Mediterranean, Gy = Guyana, AB = Argentina, SL = Sierra Leone |
||||
Fig. 9. Neighbor-joining tree comparing genetic distance in the 16S rDNA gene among individuals at abyssal and bathyal depths from both the Pacific and Atlantic. Populations were sufficiently different between bathyal and abyssal depths that they probably represent cryptic species. ATL = Atlantic, PAC = Pacific. |
||||
|
||||